Comme expliquť prťcťdemment, il est nťcessaire d'augmenter la densitť des atomes jusqu'ŗ 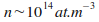 .
Pour y parvenir, la procťdure consiste en la diminution du volume contenant les atomes.
Maheureusement, dans de telles conditions, les atomes gagnent en ťnergie cinťtique et quittent le piŤge aprŤs un temps trŤs court (moins d'une seconde).
Plusieurs hypothŤses ont ťtť ťmises pour la cause de ce problŤme : il est possible que les fluctuations au cours du temps de la force dipolaire en soient ŗ l'origine.
Le but de notre stage est de rťpondre ŗ la question ę†Les fluctuations de la force dipolaire sont-elles responsables de la fuite des atomes ?†Ľ.
Pour cela, nous devons mettre au point un modŤle informatique permettant de simuler la dynamique du systŤme considťrť (atomes + piŤge) tout en ayant un total contrŰle des paramŤtres physiques.
Si nous sommes capables de concevoir une simulation suffisamment ę†rťaliste†Ľ, nous pourrons alors rťpondre ŗ la question posťe.
Il existe plusieurs facteurs influant sur le comportement des atomes au sein du piŤge (la force de pression de radiation par exemple), comme nous nous intťressons qu'ŗ seuls de ces derniers (la force dipolaire), nous ne simulerons que l'action de cette force sur les atomes.
Il n'est pas nťcessaire de simuler tous les facteurs dans la mÍme simulation pour observer les effets de l'un d'entre eux.
La dťmarche ŗ suivre est la suivante : .
Pour y parvenir, la procťdure consiste en la diminution du volume contenant les atomes.
Maheureusement, dans de telles conditions, les atomes gagnent en ťnergie cinťtique et quittent le piŤge aprŤs un temps trŤs court (moins d'une seconde).
Plusieurs hypothŤses ont ťtť ťmises pour la cause de ce problŤme : il est possible que les fluctuations au cours du temps de la force dipolaire en soient ŗ l'origine.
Le but de notre stage est de rťpondre ŗ la question ę†Les fluctuations de la force dipolaire sont-elles responsables de la fuite des atomes ?†Ľ.
Pour cela, nous devons mettre au point un modŤle informatique permettant de simuler la dynamique du systŤme considťrť (atomes + piŤge) tout en ayant un total contrŰle des paramŤtres physiques.
Si nous sommes capables de concevoir une simulation suffisamment ę†rťaliste†Ľ, nous pourrons alors rťpondre ŗ la question posťe.
Il existe plusieurs facteurs influant sur le comportement des atomes au sein du piŤge (la force de pression de radiation par exemple), comme nous nous intťressons qu'ŗ seuls de ces derniers (la force dipolaire), nous ne simulerons que l'action de cette force sur les atomes.
Il n'est pas nťcessaire de simuler tous les facteurs dans la mÍme simulation pour observer les effets de l'un d'entre eux.
La dťmarche ŗ suivre est la suivante :
- Nous simulons l'effet de la force dipolaire sur le comportement des atomes piťgťs.
- Nous quantifions le chauffage induit par cette force, c'est-ŗ-dire l'augmentation de l'energie un aprŤs un temps d'au moins une seconde
- Il se prťsente alors deux cas de figure :
-
Soit les atomes gagnent suffisamment d'ťnergie cinťtique pour quitter le piŤge aprŤs un temps semblable ou infťrieur ŗ celui obervť expťrimentalement,
on en dťduit alors que les fluctuations de la force dipolaire peuvent Ítre responsables de la fuite prťmaturťe des atomes.
-
Soit le temps de fuite des atomes est supťrieur ŗ celui observť expťrimentalement,
on en dťduit alors que les fluctuations de la force dipolaire ne sont probablement pas responsables de la fuite observťe des atomes.
- Ces prťdictions doivent Ítre vťrifiťes expťrimentalement (mais ce n'est pas le propos de notre projet)
Notre point de vue de la manipulation ne nťcessite pas de considťrer les interactions entre atomes, la simulation est donc effectuťe atome par atome, puis les rťsultats individuels sont rťunis afin de faire une satistique.
Puisque nous devons concevoir intťgralement le programme de simulation, nous commenÁons par simuler le systŤme de maniŤre trŤs simplifiťe. Nous nous plaÁons dans l'approximation de la force moyenne, dans la gťomťtrie suivante : A deux dimensions, l'atome ťvolue sur un plan repťrť par (xOy) et est piťgť entre deux murs de potentiels. Le potentiel ressenti par l'atome s'ťcrit alors
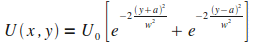
|
(1)
|
Avec 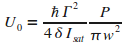
Pour se reprťsenter la gťomťtrie, on trace le graphe du potentiel.
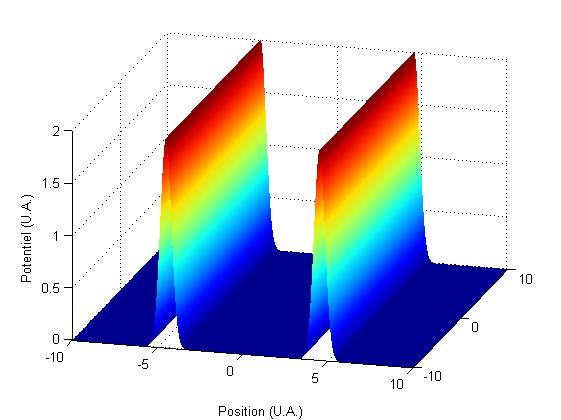
FIG. 1 - Potentiel rectiligne fixe
Pour simuler l'ťvolution temporelle de l'atome, il faut d'abord ťcrire ses ťquations du mouvement.
Pour cela on utilise le fait que la force subie par l'atome dťrive du potentiel ressenti.
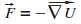
|
(2)
|
D'oý l'expression explicite de la force dipolaire.
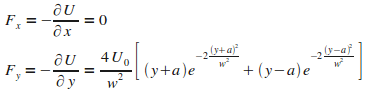
|
(3)
|
Toujours dans le but de reprťsenter la situation considťrťe, on trace le graphe de la force subie par l'atome. On remarque que cette force dťpend de la position de l'atome.
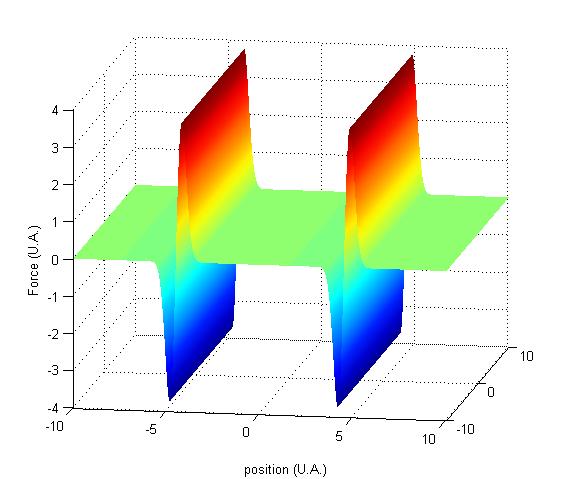
FIG. 2 - Force dipolaire dťrivant du potentiel rectiligne fixe
On ťcrit ensuite la relation fondamentale de la dynamique,
afin d'obtenir les ťquations du mouvement de l'atome en x
et en y.
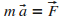
|
(4)
|
On dťcompose cette relation selon les deux composantes
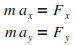
|
(5)
|
On en dťduit les ťquations du mouvement.
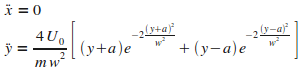
|
(6)
|
D'aprŤs ces ťquations, la particule subit une accťlťration selon la direction y,
dťpendante de la composante y de sa position,
et ne subit aucune accťlťration selon x,
ce qui se comprend car le potentiel considťrť ici est ę†rectiligne†Ľ, parallŤle ŗ l'axe x, et situť en -a et a (cf. FIG. 1).
Maintenant que nous avons les ťquations du mouvement, il faut les rťsoudre.
Nous ne pouvons bien ťvidemment pas les rťsoudre analytiquement,
c'est pourquoi nous utilisons le logiciel MatLab,
afin de rťsoudre numťriquement ces ťquations. Dans ce logiciel sont dťjŗ implťmentťs des algotrithmes de rťsolution numťrique.
Etant peu familiers avec l'utilisation de ces fonctions,
nous allons d'abord rťsoudre les ťquations diffťrentielles par la ę†mťthode d'Euler†Ľ
(il s'agit en fait d'un dťveloppement limitť ŗ l'ordre 1).
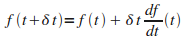
|
(7)
|
Les ťquations diffťrentielles considťrťes sont d'ordre 2, la mťthode d'Euler se fait donc en deux temps.
Il faut d'abord intťgrer les accťlťrations  et
et 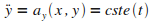 pour obtenir les vitesses
pour obtenir les vitesses 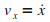 et
et 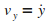 : :
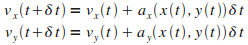
|
(8)
|
Puis intťgrer ces vitesses pour obtenir les positions en fonction du temps
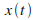 et et 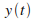 : :
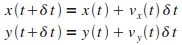
|
(9)
|
Pour appliquer cette mťthode, nous devons connaÓtre les conditions initiales 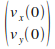 et et 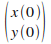 . Pour commencer nous choisissons ces conditions initiales arbitrairement, tout en restant raisonnables (c'est-ŗ-dire que nous choisissons des positions telles que l'atome soit entre les deux barriŤres de potentiel, avec une vitesse de l'ordre du m/s). . Pour commencer nous choisissons ces conditions initiales arbitrairement, tout en restant raisonnables (c'est-ŗ-dire que nous choisissons des positions telles que l'atome soit entre les deux barriŤres de potentiel, avec une vitesse de l'ordre du m/s).
L'algorithme d'Euler se dťroule alors ainsi :
|
Initialisation
|
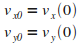
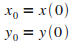
|
|
Itťration n = 1
|
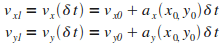
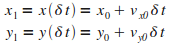
|
|
Itťration n = 2
|
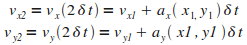
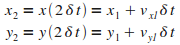
|
|
Itťration n = 3
|
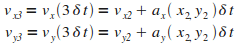
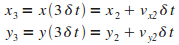
|
|
|

|
|
Itťration n
|
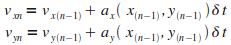
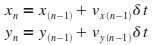
|
Il faut maintenant choisir un pas de simulation 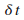 .
Pour cet essai, nous choisissons un pas arbitraire, relativement petit par rapport ŗ la seconde,
de l'ordre de 100Ķs, dans le but d'avoir une rťsolution assez prťcise.
Maintenant, nous pouvons traduire cet algorithme en langage " MatLab ".
En le plaÁant dans une boucle for,
on obtient alors la position .
Pour cet essai, nous choisissons un pas arbitraire, relativement petit par rapport ŗ la seconde,
de l'ordre de 100Ķs, dans le but d'avoir une rťsolution assez prťcise.
Maintenant, nous pouvons traduire cet algorithme en langage " MatLab ".
En le plaÁant dans une boucle for,
on obtient alors la position 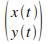 au cours du temps jusqu'ŗ
au cours du temps jusqu'ŗ 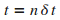 . .
Dans cette gťomťtrie, l'accťlťration selon x est nulle,
on constate donc que la vitesse selon cette direction reste constante,
tandis que la vitesse selon y subit l'accťlration
dans la direction y.
Pour observer le rťsultat, on trace la trajectoire de l'atome :
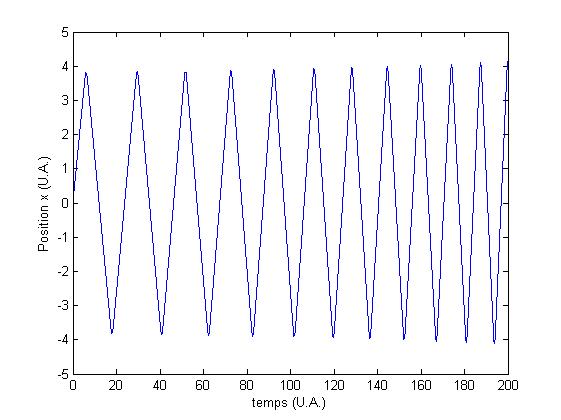
FIG. 3 - Trajectoire dans un potentiel rectiligne
Afin de se familiariser davantage avec l'utilisation de MatLab, nous effectuons la mÍme simulation, avec une gťomťtrie plus proche de la rťalitť de l'expťrience. Nous sommes toujours ŗ 2 Dimensions, dans l'approximation de la force moyenne, mais cette fois-ci l'atome est entourť d'un potentiel circulaire de rayon a.
Cette approximation peut se justifier ainsi : La frťquence de rotation des faisceaux lasers qui piŤgent l'atome (cf. rapport + introduction) est tellement ťlevťe qu'ils forment une "cage" autour de l'atome, constante au cours du temps. Si l'atome ne se dťplace que sur 2 dimensions, cette "cage" se modťlise par un "mur" circulaire.
Le potentiel considťrť s'exprime alors :
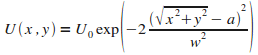
|
(10)
|
On trace son graphe :
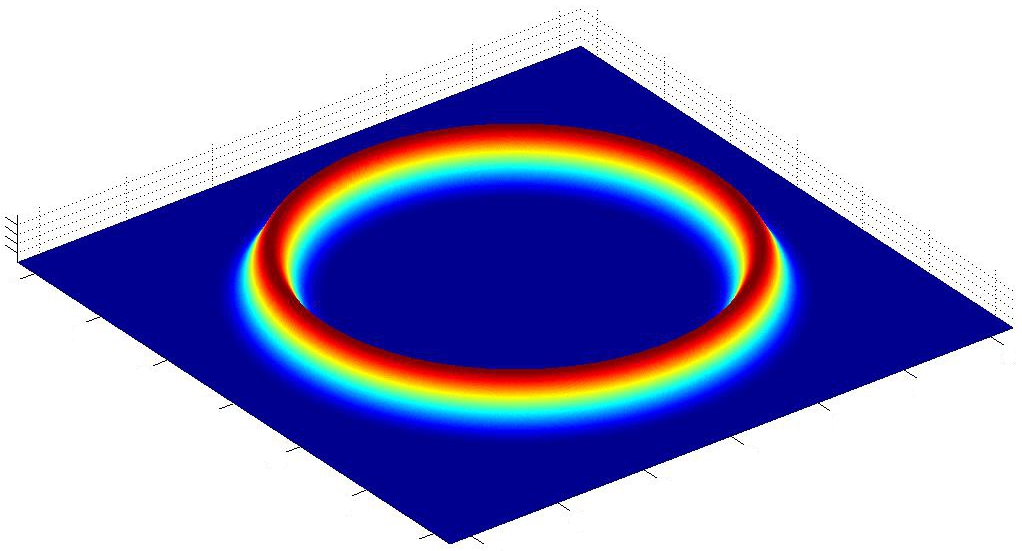
FIG. 4 - Potentiel circulaire fixe
En adoptant la mÍme dťmarche que prťcťdemment
(on exprime la force dipolaire avec le gradient du potentiel, puis on ťcrit le relation fondamentale de la dynamique),
on obtient les ťquations du mouvement (et donc les accťlťrations) :
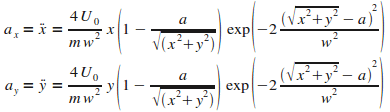
|
(11)
|
Maintenant que nous avons les accťlťrations selon x et y pour cette gťomťtrie, nous procťdons exactement de la mÍme faÁon que pour la gťomťtrie prťcťdente, et nous obtenons donc la position de l'atome au cours du temps.
On peut alors tracer la trajectoire de l'atome
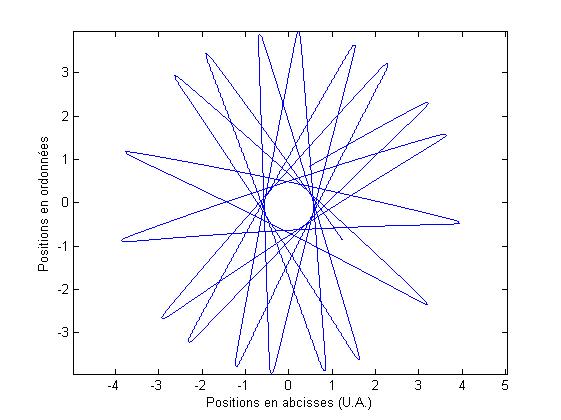
FIG. 5 - Trajectoire dans le piŤge circulaire fixe
D'aprŤs le potentiel que nous avons choisi, il semble que la trajectoire rťsolue numťriquement soit correcte.
Nous sommes, pour l'instant, suffisamment familiers avec la rťsolution numťrique d'ťquations diffťrentielles,
nous allons donc nous intťresser ŗ l'objet de notre stage.
Notre but est d'ťtudier l'effet des fluctuations de la force dipolaire.
Nous ťtudions le systŤme physique ŗ 2 dimensions, afin de simplifier le code de la simulation.
Il s'agit d'ťtudier l'interaction d'un atome avec un potentiel mais, cette fois-ci,
le potentiel considťrť dťpend du temps :
c'est une gaussienne ŗ 2 dimensions en rotation ŗ une frťquence
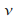 autour d'un point (que l'on choisit ŗ l'origine du repŤre),
dťcrivant un cercle de rayon a.
Ce potentiel rotatif est la modťlisation du faisceau laser oscillant permettant de piťger les atomes
(cf. rapport + introduction).
Si on fait une moyenne temporelle de ce potentiel tournant, on retrouve la configuration prťcťdente,
avec un potentiel circulaire. autour d'un point (que l'on choisit ŗ l'origine du repŤre),
dťcrivant un cercle de rayon a.
Ce potentiel rotatif est la modťlisation du faisceau laser oscillant permettant de piťger les atomes
(cf. rapport + introduction).
Si on fait une moyenne temporelle de ce potentiel tournant, on retrouve la configuration prťcťdente,
avec un potentiel circulaire.
L'expression du potentiel dťpendant du temps est la suivante :
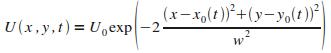
|
(12)
|
Avec
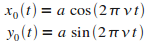
|
(13)
|
A un instant donnť le potentiel ressemble alors ŗ Áa :
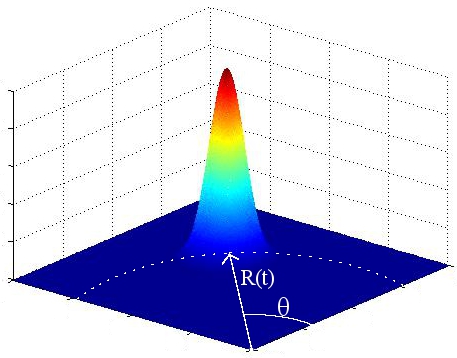
FIG. 6 - Potentiel circulaire tournant, ŗ un instant donnť
On procŤde de la mÍme faÁon que prťcťdemment pour ťcrire les ťquations du mouvement de l'atome dans cette configuration. On obtient :
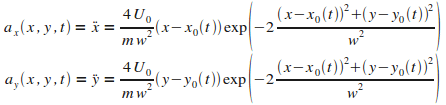
|
(14)
|
On remarque que, cette fois-ci, contrairement aux cas prťcťdents,
les accťlťrations selons les deux composantes dťpendent du temps.
Cela pose alors un nouveau problŤme :
le pas de simulation ne peut plus Ítre choisi arbitrairement.
En effet, le potentiel est une gaussienne tournante ŗ la vitesse angulaire
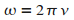 ,
pour simuler la manipulation de maniŤre rťaliste, le dťplacement du potentiel au cours du temps doit Ítre continu.
Autrement dit, entre deux itťrations de la simulation, le potentiel ne doit pas faire de " saut ",
sans quoi l'atome pourrait intempestivement quitter le piŤge.
Pour rťaliser cette condition, on choisit le pas de simulation tel que,
pendant ,
le potentiel se dťplace de 1/20e de sa largeur w.
D'aprŤs les equations (12) et (13),
on voit que, pendant un pas de simulation,
le potentiel se dťplace d'un angle ,
pour simuler la manipulation de maniŤre rťaliste, le dťplacement du potentiel au cours du temps doit Ítre continu.
Autrement dit, entre deux itťrations de la simulation, le potentiel ne doit pas faire de " saut ",
sans quoi l'atome pourrait intempestivement quitter le piŤge.
Pour rťaliser cette condition, on choisit le pas de simulation tel que,
pendant ,
le potentiel se dťplace de 1/20e de sa largeur w.
D'aprŤs les equations (12) et (13),
on voit que, pendant un pas de simulation,
le potentiel se dťplace d'un angle 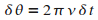 ,
et donc d'un distance ,
et donc d'un distance 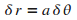 .
On impose la valeur de cette distance .
On impose la valeur de cette distance 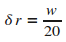 .
On en dťduit la valeur du pas de simulation : .
On en dťduit la valeur du pas de simulation :
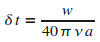
|
(15)
|
Nous avons maintenant un pas de simulation convenable,
nous pouvons donc rťsoudre numťriquement les ťquations du mouvement, comme nous l'avons fait prťcťdemment.
Nous obtenons alors la trajectoire de l'atome :
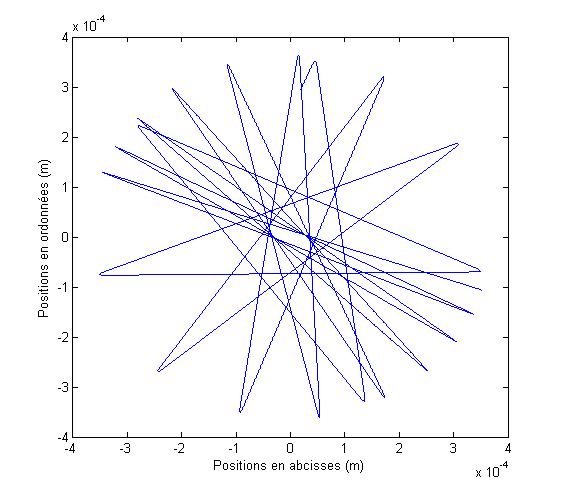
FIG. 7 - Trajectoire dans le potentiel dipolaire dťpendant du temps
Visuellement, il semble que l'on observe bien un atome piťgť dans un potentiel tournant.
Maintenant, nous dťcidons de faire une simulation avec les paramŤtres physiques de la manipulation.
Il convient alors de dťfinir les constantes dans le code.
Dans le but d'obtenir une rťsolution la plus prťcise possible, nous n'utilisons dorťnavant plus la "mťthode d'Euler",
mais nous utilisons l'algorithme de Runge-Kutta d'ordre 4. Nous connaissons cet algorithme en langage C,
nous l'adaptons nous-mÍme ŗ MatLab.
Nous ne le dťtaillerons pas ici,
car nous nous apercevrons finalement qu'il induit une dťviation numťrique trop ťlevťe pour observer un comportement
physiquement crťdible (voir plus loin).
Comme pour la mťthode d'Euler, nous devons dťfinir un pas de simulation.
Nous utilisons donc le calcul expliquť prťcťdemment.
Dans les conditions physiques de la manipulation,
nous obtenons 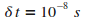 . .
Nous effectuons donc la simulation pour un atome, puis nous traÁons sa trajectoire.
Nous obtenons un rťsultat similaire ŗ la trajectoire prťcťdente,
ce qui peut nous laisser croire que notre algorithme de Runge-Kutta fonctionne correctement.
Dans le cadre de notre ťtude, nous devons regarder l'ťnergie cinťtique des particules.
Nous n'avons, ŗ partir de maintenant, plus besoin de tracer de trajectoires.
La mťthode Runge-Kutta, tout comme la mťthode d'Euler, se fait en deux temps (intťgration des accťlťrations pour obtenir les vitesses, puis intťgration des vitesses pour obtenir les coordonnťes).
La rťsolution numťrique nous donne alors les vitesses et les positions ŗ chaque instant.
Il est donc possible d'ťcrire l'ťnergie cinťtique des atomes ŗ chaque instant.
Ainsi, nous pouvons observer son ťvolution temporelle, puis quantifier le chauffage induit par les fluctuations force dipolaire.
Sachant que les ťnergies cinťtiques mises en jeu sont de l'ordre de 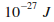 ,
pour ne pas dťpasser la prťcision de l'ordinateur, nous comptons l'ťnergie cinťtique en Kelvins.
Pour cela nous divisons l'ťnergie par la constante de Boltzmann k. ,
pour ne pas dťpasser la prťcision de l'ordinateur, nous comptons l'ťnergie cinťtique en Kelvins.
Pour cela nous divisons l'ťnergie par la constante de Boltzmann k.
A l'aide des coordonnťes ( x(t), y(t) ), nous sommes capables de dťtecter un atome qui quitterait le piŤge,
et de vťrifier, avec l'ťnergie cinťtique,
que cette fuite provient effectivement d'une trop grande ťnergie de l'atome (qu'elle a donc chauffť) et non d'une erreur numťrique.
Pour cela on compare l'ťnergie de l'atome ŗ l'instant de sa sortie avec la hauteur du potentiel moyen (de l'ordre de 1000 ĶK).
Forts de cet outil numťrique, nous effectuons la simulation sur 1000 atomes. Au bout de 7 jours, seulement 49 atomes ont ťtť simulťs.
En faisant une moyenne sur ces atomes, nous traÁons l'ťnergie cinťtique par atome.
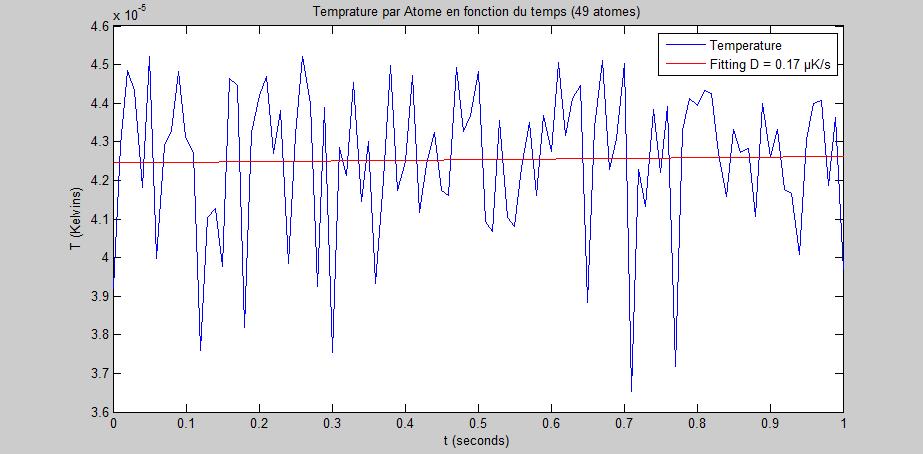
FIG. 8 - Energie cinťtique par atome au cours du temps
Nous observons que le chauffage peut Ítre considťrť comme absent. Or, la thťorie prťdit que le chauffage doit Ítre prťsent.
Nous rťalisons d'autres simulations, et observons les rťsultats, dont voici un ťchantillon.
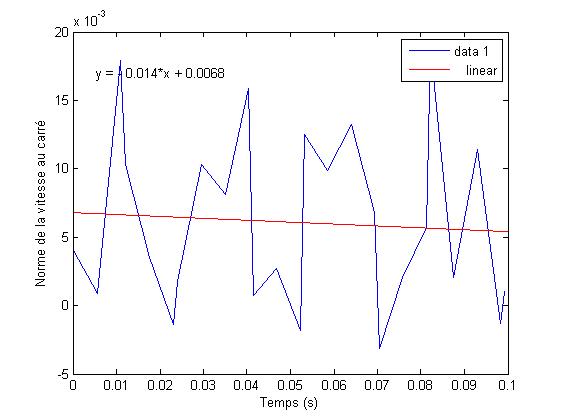
FIG. 9 - Energie cinťtique par atome au cours du temps
Ici on observe mÍme un refroidissement, ce qui est physiquement impossible.
Nous avons pris beaucoup de temps (environ les 4/5e du temps restant pour le projet tutorť) ŗ essayer de modifier le code pour obtenir des rťsultats plus cohťrents,
nous avons par exemple conÁu un pas de simulation adaptatif
(dans la zone centrale du piŤge, il n'y pas d'interactions, on peut alors augmenter le pas de temps,
tandis qu'au voisinage du cercle dťcrit par le faisceau laser, les interactions entre atome et faisceau laser sont nombreuses,
le pas doit donc Ítre le plus court possible).
Finalement, nous en avons conclu que notre algorithme de Runge-Kutta d'ordre 4 n'est pas assez prťcis,
la dťviation numťrique qu'il induit est trop ťlevťe pour un tel problŤme physique.
Nous dťcidons alors d'apprendre ŗ utiliser les fonctions de rťsolution numťrique dťjŗ implťmentťes dans MatLab
(par exemple ode45 ou ode15s).
Notre systŤme d'ťquations diffťrentielles est d'ordre 2.
Pour utiliser les fonctions de rťsolution de MatLab, il faut dťcomposer ce systŤme d'ťquations en un systŤme d'ťquations d'ordre 1 :
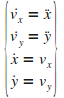
|
(16)
|
Nous traduisons ce systŤme en langage MatLab, afin que les fonctions de rťsolution puissent le rťsoudre :
Il faut dťfinir un vecteur X_dot = [ acc_x, acc_y, vx, vy ]
contentant les dťrivťes temporelles de chaque composante du systŤme d'ťquations .
Et un vecteur X = [ vx, vy, x, y ] contenant les fonctions ŗ rťsoudre.
Il suffit alors d'entrer cette commande dans MatLab
[t X] = ode15s( @acc, [0 tfin], [ vxini, vyini, xini, yini ], odeset( 'RelTol', 2.22045e-014, 'MaxStep', 1e-6, 'InitialStep', 1e-8 ) );
L'argument @acc appelle la fonction qui dťfinit X_dot.
Le vecteur [ vxini, vyini, x,ini, yini ] contenant les conditions initiales.
La commande odeset( 'RelTol', 2.22045e-014, 'MaxStep', 1e-6, 'InitialStep', 1e-8 ) est nťcessaire.
Elle permet:
- De rťduire au minimum la tolťrance relative de l'erreur ŗ chaque pas de simulation.
-
De fixer un pas de simulation maximal permettant ŗ l'atome de ne pas faire de trop grand "saut",
mÍme quand le pas est maximal, sans quoi il risquerait de quitter le piŤge
- De fixer un pas de simulation initial convenable, sans quoi la rťsolution numťrique est totalement fausse.
Nous obtenons alors un vecteur t contenant le temps ŗ chaque pas de simulation,
et une matrice X composťe de 4 colonnes contenant, respectivement,
les vitesses en x et y,
puis les coordonnťes x et y, ŗ chaque pas de simulation.
Nous rťalisons alors une simulation sur 390 atomes (au bout de 7 jours),
et enregistrons le temps, les vitesses et les coordonnťes de chaque atome dans un fichier
(en ťcrivant un fichier par atome).
Tout comme prťcťdemment, nous observons l'ťnergie cinťtique des atomes au cours du temps.
Cette fois-ci, nous observons effectivement un chauffage induit par la force dipolaire.
Nous pouvons alors le quantifier et en tirer les conclusions dans la partie exploitation.
|

